Tumor vs Normal Classification Using Deep Learning

Understanding how the immune system interacts with cancer is a hot topic in bioinformatics. In this project, I explore how T-cell receptor (TCR) CDR3 sequences—which capture a patient’s immune landscape—can help distinguish tumor from normal samples using deep learning.
TL;DR: I built a pipeline that takes TRB CDR3 sequences, tokenizes them, trains a simple deep learning model (Mean Pooling & LSTM), and evaluates classification performance. Spoiler: it’s harder than it looks.
Motivation
TCR CDR3 sequences represent how T-cells recognize antigens. Since tumors interact with the immune system in complex ways, my hypothesis was:
“Can we classify tumor vs normal samples based on their repertoire of TRB CDR3 sequences?”
This is both biologically meaningful and a great chance to practice deep learning on non-traditional text-like biological data.
Preprocessing CDR3 Data
I started with two CSV files:
- One for tumor samples
- One for normal samples
These cancer files were obtained from the GDC CPTAC Lung cohort and include both adenocarcinoma and squamous cell carcinoma types combined.
Each row was a CDR3 sequence with a Sample ID field.
1
2
3
4
5
6
7
# Load and label data
df_tumor = pd.read_csv(tumor_file)
df_normal = pd.read_csv(normal_file)
df_tumor["label"] = "tumor"
df_normal["label"] = "normal"
df_all = pd.concat([df_tumor, df_normal])
CDR3 per patient
To build patient-level models, I grouped CDR3s by Sample ID.
1
cdr3_counts = df_all.groupby("Sample ID")["CDR3"].nunique()
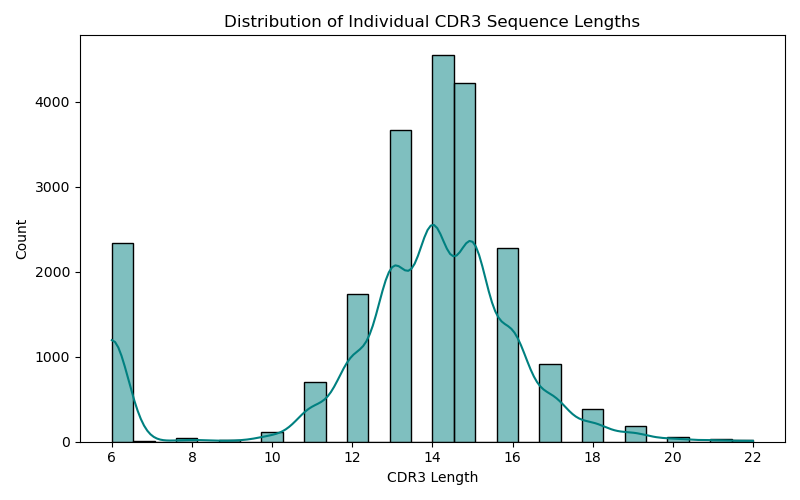
Filtering and Tokenization
Filter patients with enough sequences
To avoid noise, I only kept patients with ≥20 unique CDR3s. Then I normalized to exactly 20 sequences per patient via:
- Padding (with
"PADSEQ") if <20 - Random sampling if >20
1
2
MAX_CDR3S = 20
PADSEQ = "PADSEQ"
Tokenizing amino acids
Each CDR3 sequence (e.g., 'CASSLGQGAETQYF') was tokenized using an amino acid vocabulary.
1
2
3
4
5
6
7
8
AMINO_ACIDS = list("ACDEFGHIKLMNPQRSTVWY")
VOCAB = {aa: i+2 for i, aa in enumerate(AMINO_ACIDS)}
VOCAB["<PAD>"] = 0
VOCAB["<UNK>"] = 1
def tokenize_sequence(seq, max_len=30):
tokens = [VOCAB.get(aa, VOCAB["<UNK>"]) for aa in seq]
return tokens[:max_len] + [VOCAB["<PAD>"]] * (max_len - len(tokens))
Each patient becomes a 20×30 matrix (20 CDR3s of length 30, padded if needed)
Building the Deep Learning Model
Model 1: Mean Pooling Baseline
This model embedded each amino acid, averaged the 20 CDR3 embeddings, and passed it through a classifier.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
class MeanPoolModel(nn.Module):
def __init__(self, vocab_size, embed_dim):
super().__init__()
self.embedding = nn.Embedding(vocab_size, embed_dim, padding_idx=0)
self.classifier = nn.Sequential(
nn.Linear(embed_dim, 64),
nn.ReLU(),
nn.Linear(64, 1),
nn.Sigmoid()
)
def forward(self, x):
# x shape: [batch, 20, 30]
embedded = self.embedding(x) # [batch, 20, 30, embed_dim]
mean_emb = embedded.mean(dim=[1, 2]) # Mean pooling
return self.classifier(mean_emb).squeeze()
Evaluation
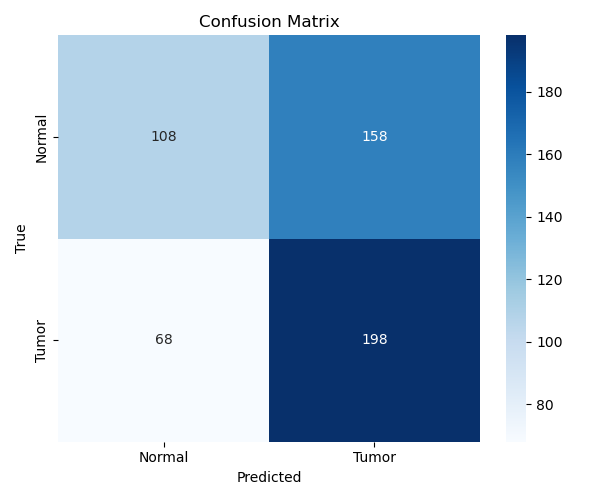
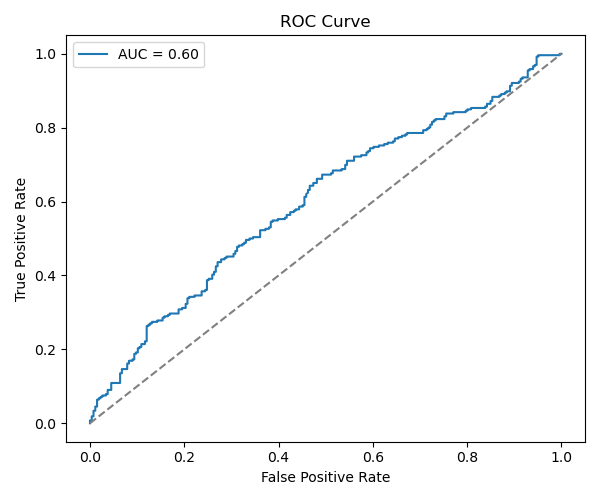
Performance
1
2
3
Accuracy: ~58%
F1 Score (Tumor): 0.64
F1 Score (Normal): 0.49
Model 2: LSTM Attempt
Instead of mean pooling, I passed each patient’s 20×30 matrix through an LSTM and used the final hidden state for classification.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
class LSTMClassifier(nn.Module):
def __init__(self, vocab_size, embed_dim, hidden_dim):
super().__init__()
self.embedding = nn.Embedding(vocab_size, embed_dim, padding_idx=0)
self.lstm = nn.LSTM(embed_dim, hidden_dim, batch_first=True)
self.fc = nn.Linear(hidden_dim, 1)
self.sigmoid = nn.Sigmoid()
def forward(self, x):
batch_size, num_seqs, seq_len = x.size()
x = x.view(batch_size * num_seqs, seq_len)
emb = self.embedding(x)
_, (h_n, _) = self.lstm(emb)
h_n = h_n.squeeze()
h_n = h_n.view(batch_size, num_seqs, -1).mean(dim=1)
return self.sigmoid(self.fc(h_n)).squeeze()
Result? Still poor generalization. Slightly better F1 but no significant boost.
Limitations & Future Directions
Although this project successfully walked through every step of building a deep learning model on TRB CDR3 sequences—from raw CSVs to model evaluation—the final performance didn’t meet expectations. There are several likely reasons. First, the biological differences in TRB repertoires between tumor and normal tissues may not be dramatic or easily captured at the global level, especially when only amino acid sequences are used without any V/D/J gene context. Second, the models I implemented—mean pooling and LSTM—were relatively simple. While they helped me learn foundational DL techniques like embeddings, padding, and sequential modeling, they may have been insufficient to capture more nuanced patterns or structural motifs present in the data. Also, padding all CDR3s to a fixed length could have diluted signal in shorter sequences, which might be biologically relevant.
Going forward, I plan to improve this baseline by exploring more advanced architectures such as attention-based models, which may be better suited to highlight critical CDR3s within a repertoire. Adding V/D/J gene features, using pre-trained amino acid embeddings, or applying transfer learning are promising directions. I also want to try unsupervised clustering to explore whether natural grouping of repertoires exists before forcing a supervised classifier on the data. Importantly, obtaining more balanced and high-quality normal samples will likely make a big difference. These improvements will be made iteratively as I continue learning and experimenting with deep learning in immunogenomics.
Final Thoughts
This was a fantastic learning experience! Even though performance wasn’t great, the project showed how TCR repertoire data can be used in DL pipelines, and revealed how challenging immune modeling is.
Will I do more immune-seq modeling? Absolutely. With attention mechanisms, pretraining, or maybe a better dataset. Stay tuned!
Full code available at - Github